2024年8月09日
2024年8月09日
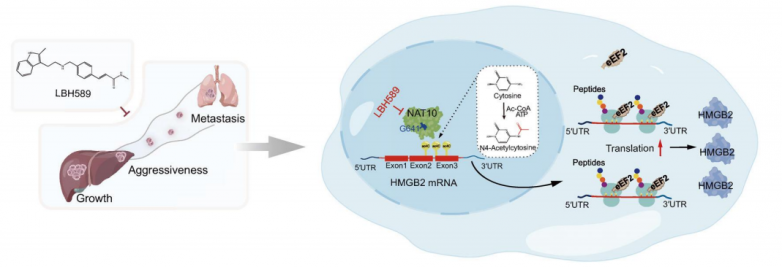
肝细胞癌(HCC)是全球第四大癌症死亡原因,致死率极高。当前针对晚期HCC的治疗方法,主要靶向肿瘤免疫微环境(TIME),而非直接针对肿瘤细胞。前期的研究表明异常的ac4C(N4-乙酰胞苷)修饰与多种实体瘤的进展有关,但是,ac4C在HCC进展中明确的生物学意义和关键的机制仍然是个谜。2024年7月,武汉同济医院张必翔团队一篇题为“Targeting N4-acetylcytidine suppresses hepatocellular carcinoma progression by repressing eEF2-mediated HMGB2 mRNA translation”的研究成果发表在Cancer Communications期刊上,该研究揭示了NAT10-ac4C/eEF2-HMGB2表观转录组轴在调节HCC生长和转移中的关键作用,且验证了帕比司他(Panobinostat)是针对NAT10介导的ac4C的一种有效且安全的先导化合物,具有潜在的肝癌治疗价值。
迈维代谢为该研究提供了RNA甲基化靶向代谢组学检测!

研究方法
组学方法:ac4C靶向检测、转录组测序、acRIP-seq、DIA定量蛋白组检测等;
其他方法:qRT-PCR、Western blot分析、细胞培养、转染和病毒转导、免疫组化(IHC)检测等;
研究结果
1.RNA ac4C高乙酰化和NAT10上调与肝癌预后不良相关
为了阐明ac4C修饰在肝细胞癌(HCC)中的功能作用,研究人员检测了20例HCC组织和癌旁组织中RNA ac4C 的水平。点印迹(Dot blot)实验显示,HCC组织总RNA中ac4C水平显著升高(图1A)。这一发现通过超高效液相色谱-串联质谱(UPLC-MS/MS)RNA甲基化靶向代谢组分析进一步得到证实,该分析显示HCC组织mRNA中ac4C水平显著升高(图1B)。比较了20对HCC和正常肝组织中NAT10(负责ac4C修饰的唯一乙酰转移酶)的mRNA水平,结果表明NAT10在HCC中显著上调,并且在20例HCC组织中NAT10表达与 RNA ac4C水平之间存在显著相关性。为了进一步验证这些结果,研究人员对来自89组HCC患者的数据进行了大规模数据挖掘分析。其中62个队列内mRNA NAT10 水平有显著变化。54个队列HCC组织中mRNA NAT10 水平显著升高(图1C)。接下来,使用来自临床蛋白质组学肿瘤分析联盟(CPTAC)的数据集进行了基因表达分析,该数据集支持NAT10在HCC中的上调(图1D)。与这些发现一致,通过蛋白质印迹法分析,人HCC组织中NAT10的蛋白质水平显著高于癌旁组织(图1E)。对来自HCC组织微阵列(同济队列)的103对样本的免疫组织化学分析,进一步证实了HCC样本中NAT10的上调(图1F)。此外,NAT10的高表达与HCC患者的总生存期和无复发生存期显著缩短有关(图1G)。
后续分析表明,NAT10高表达与患者HCC组织和与不良预后相关的分子亚型iCluster-1 HCC样本中的临床晚期显著相关。NAT10表达在HCC发展过程中逐渐增加(图1H)。综上所述,这些发现表明NAT10表达与HCC的恶性进展密切相关。
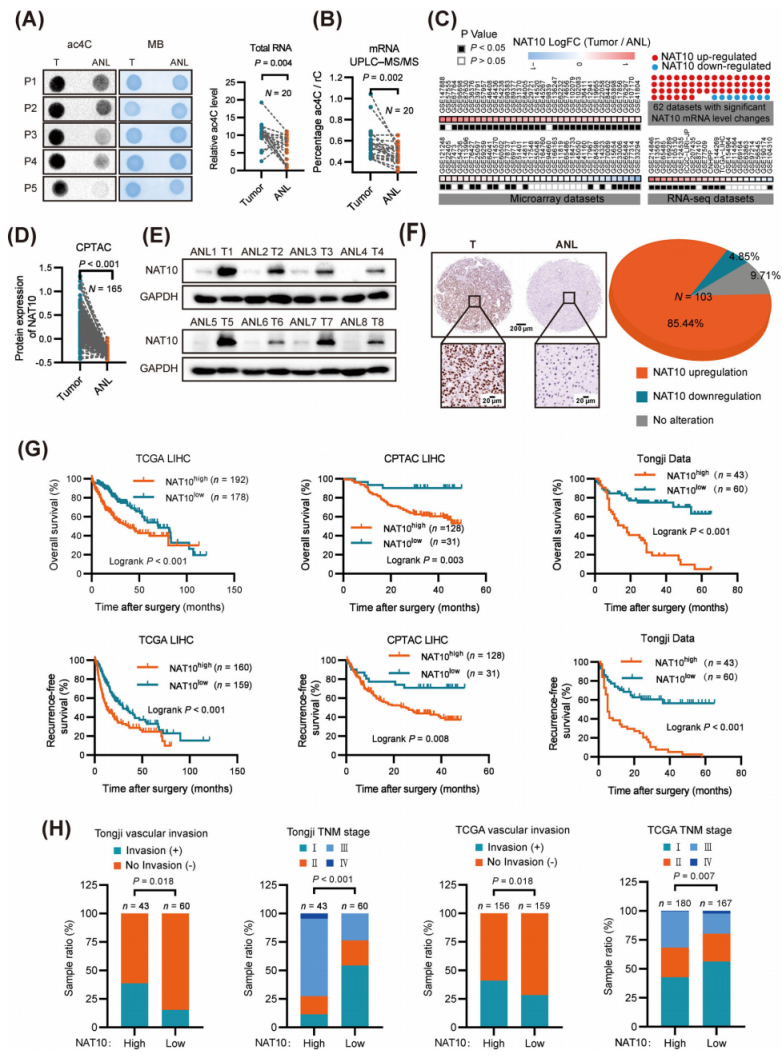
图1.在HCC患者中,NAT10表达升高与预后不良相关
2.下调NAT10可抑制HCC的增殖和转移
为了研究NAT10在肝细胞癌(HCC)中的功能,研究人员使用NAT10表达和ac4C修饰水平均升高的细胞系(MHCC-97H和SNU449)建立了稳定的NAT10敲低细胞系。与对照shRNA(shCtrl)相比,NAT10的敲低显著降低了总RNA和mRNA中的ac4C含量(图2A)。NAT10沉默显著抑制了HCC细胞的活力(图2B-C)、克隆形成能力(图2D)以及软琼脂菌落形成效率(图2E)。此外,伤口愈合迁移、Transwell迁移和基质胶侵袭实验表明,NAT10沉默显著抑制了MHCC-97H和SNU449细胞的迁移和侵袭能力(图2F-G)。随后,研究人员评估了NAT10在体内对HCC增殖和转移的影响。肿瘤异种移植研究表明,NAT10沉默显著抑制了肿瘤生长,表现为与对照细胞来源的肿瘤相比,肿瘤大小和重量减少(图2H)。此外,肿瘤异种移植中NAT10的敲低导致细胞增殖受到抑制并诱导凋亡,这分别通过Ki-67和末端脱氧核苷酸转移酶介导的脱氧尿苷三磷酸缺口末端标记(TUNEL)染色得到证实(图2I-J)。此外,在肝脏原位植入模型中,与对照组相比,接种NAT10敲低细胞的组肝内肿瘤结节的体积和数量显著减少(图2K)。此外,在通过向裸鼠的侧尾静脉注射NAT10敲低细胞和阴性对照细胞建立的肺转移模型中,NAT10沉默显著抑制了HCC的肺转移(图2L)。综上所述,这些发现表明NAT10在HCC的增殖和转移中作为致癌驱动因子发挥着关键作用。
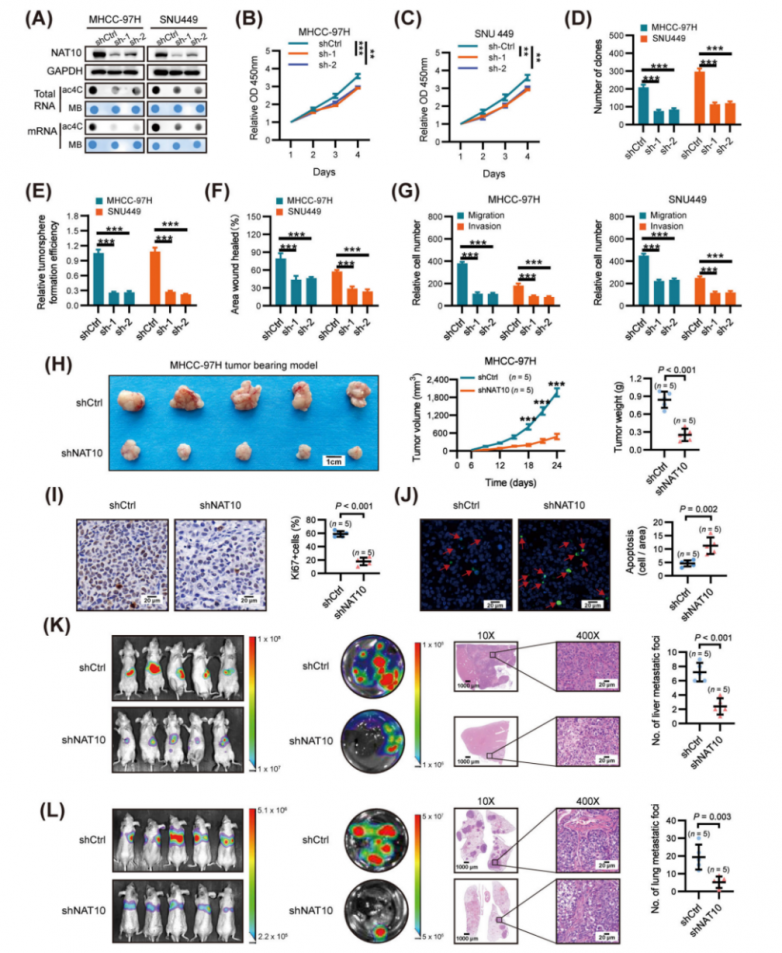
图2.在体外和体内,NAT10敲低抑制HCC进展
3.NAT10对mRNA ac4C修饰和全局mRNA翻译的影响
分别对NAT10敲除和不敲除的MHCC-97H细胞中进行了acRIP-seq实验(图3A)。确定了对照组细胞中来自4784个ac4C修饰的转录本的共11591个ac4C峰,以及NAT10缺陷细胞中来自691个ac4C修饰的转录本的3148个峰(图3B-C)。值得注意的是,在稳定的NAT10敲除细胞中出现了609个新的峰,而9052个峰消失了(图3B)。由于NAT10是一种ac4C乙酰转移酶,因此这9052个独特的峰预计包含NAT10的真正靶标。在检测到的峰中富集了ac4C共有基序“CXX”(图3D),这与先前的研究结果一致。随后,研究人员比较了有或没有shNAT10的HCC细胞中的acRIP-seq数据,分析了差异ac4C峰及其对应的转录本。在shNAT10细胞中鉴定出125个转录本显示出ac4C峰减少(图3F),这些结果共同表明NAT10在HCC中介导ac4C mRNA乙酰化。通过RNAseq和Ribo-seq实验,鉴定受NAT10介导的ac4C修饰影响的下游功能效应子(图3G)。NAT10的耗竭极大地改变了基因转录表达和核糖体保护片段(RPF)的丰度(图3H)。从RNA-seq中,在NAT10耗竭后检测到635个下调和818个上调的转录本。从Ribo-Seq中,NAT10敲低后鉴定出277个下调和332个上调的转录本。mRNAs ac4C(-)在响应NAT10缺失时显示出特定的T.E(翻译效率)升高(图3I)。多聚体分析显示,在NAT10缺陷细胞中,80S单体和多聚体的组装显著减少(图3J),表明NAT10的耗竭抑制了全局蛋白质翻译。此外,使用SUnSET试验测定时,NAT10的耗竭显著抑制了蛋白质合成(图3K)。所有这些结果表明NAT10介导的mRNA ac4C修饰增强了肝细胞癌中的全局翻译效率。
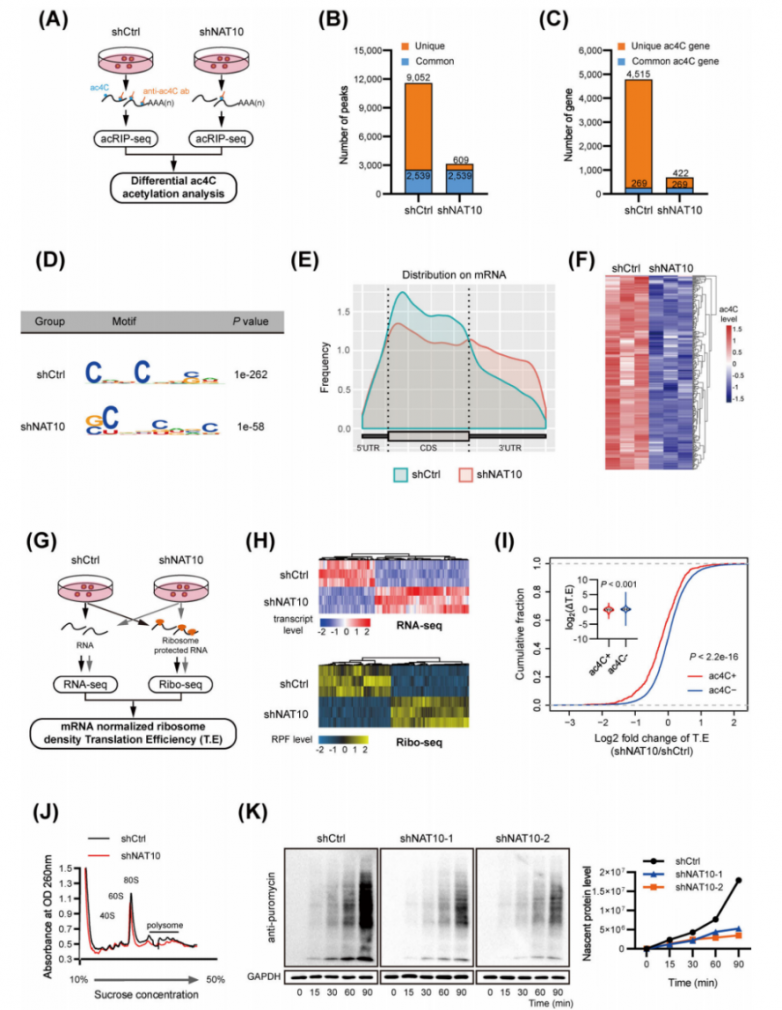
图3.NAT10对mRNA ac4c修饰和全局mRNA翻译的影响
4.NAT10介导的ac4C修饰增强HMGB2 mRNA的翻译
研究人员将来自acRIP-seq、RNA-seq和Ribo-seq的重叠候选基因进行重叠,以精确定位下游靶标(图4A)。在潜在的NAT10下游靶标(HMGB2、HIF0、PITX1和STC2)中,HMGB2在NAT10耗竭后显示出最显著的下调(图4B-C),这表明HMGB2可能是HCC细胞中NAT10的直接靶标。acRIP-seq和Ribo-seq数据显示,在NAT10缺陷细胞中,CDS中的ac4C富集和核糖体占据率显著降低(图4D)。相应地,NAT10敲低显著降低了HMGB2 mRNA的ac4C水平和蛋白质表达(图4E-F),而其mRNA丰度未受影响。有趣的是,使用MG132抑制蛋白酶体未能恢复NAT10沉默的HCC细胞中HMGB2的蛋白质表达(图4G),这表明NAT10诱导的HMGB2表达不受蛋白质降解变化的影响。将HMGB2 CDS连接到多克隆位点(MCS)来构建了pmirGLO-HMGB2荧光素酶报告基因,双荧光素酶测定清楚地表明,与对照细胞相比,NAT10缺陷细胞中HMGB2的翻译效率显著降低(图4H,补充图S4B-C)。此外,与对照细胞相比,NAT10缺陷细胞中HMGB2 mRNA在核糖体组分中的分布显示翻译活性多聚体(> 80S)显著减少(图4I)。这些综合结果表明,ac4C诱导的HMGB2表达与翻译调控有关。
在NAT10敲低细胞中过表达HMGB2显著恢复了细胞的体外增殖和侵袭能力。此外,体内实验表明,HMGB2完全挽救了NAT10敲低后的异种移植瘤生长和肺转移(图4J-L)。该数据表明,HMGB2是NAT10的关键功能性靶标。此外,在同济医院和CPTAC队列中分析了HMGB2的临床相关性,揭示了HCC组织中HMGB2的表达升高。相关性分析进一步揭示了HCC中高表达的HMGB2与NAT10水平之间存在显著关联(图4M-N)。此外,生存分析表明,这两个基因的高表达均预示着最差的总体生存期(OS)和无复发生存期(RFS)。综上所述,NAT10介导的ac4C修饰通过增强HMGB2的翻译促进了HCC的恶性进展。
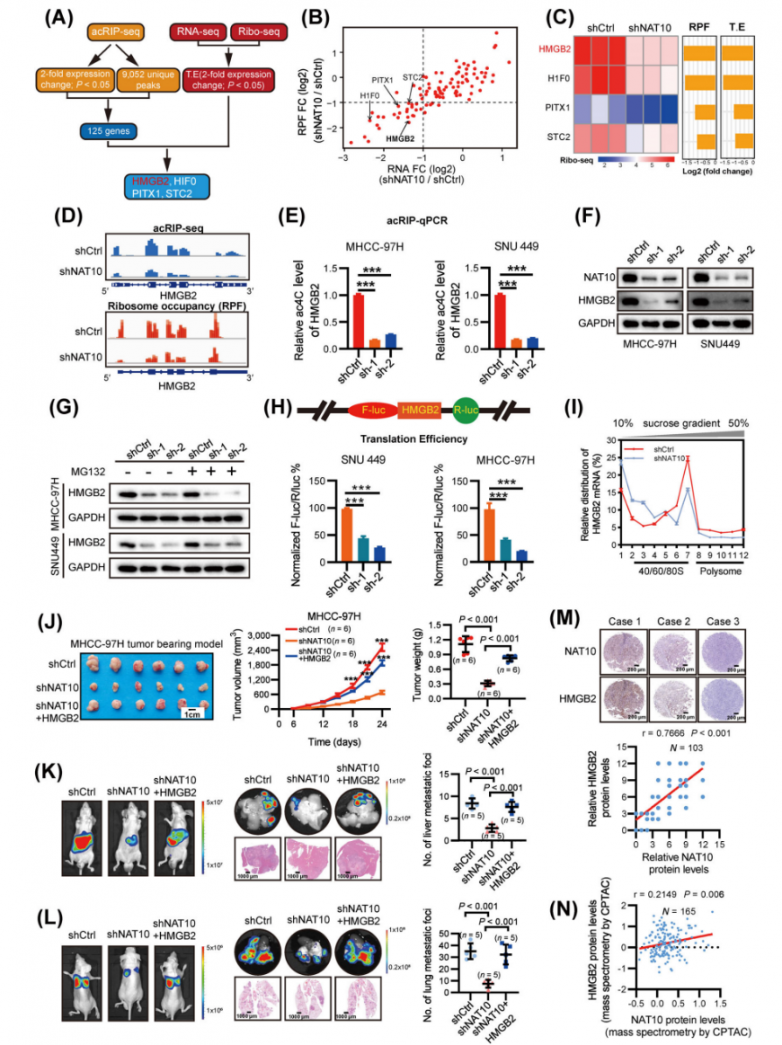
图4.ac4C的修饰增强了HMGB2的翻译
5.HMGB2 mRNA的CDS ac4C位点增强eEF2的结合
RNA的乙酰化也可能影响相互作用蛋白的结合,这与特异性结合蛋白通过介导mRNA翻译来识别RNA中的N6-甲基腺苷相似。为了鉴定直接结合ac4C修饰的蛋白质,研究人员使用含有或不含ac4C的生物素标记寡核苷酸进行了RNA亲和色谱法和数据非依赖性采集(DIA)蛋白质组学定量检测(图5A-B)。结果,在ss-ac4C组中,检测到123种蛋白质。GO分析表明,蛋白质主要富集在翻译延伸和翻译等方面(图5C)。此外,还在STRING蛋白质-蛋白质相互作用(PPI)数据库中搜索了这些鉴定出的蛋白质,以查找已知的PPI,并根据通过GO分析获得的已知功能对蛋白质节点进行了分组。根据这些蛋白质已知的生物学功能,将它们分为六个主要亚群,包括翻译、靶向内质网、核糖核蛋白等。所有这些结果都表明,ac4C修饰参与了翻译过程。对ss-ac4C和acFUS结合蛋白进行重叠分析,鉴定出了eEF2和SRP68(图5D-E)。RNA下拉实验证实,eEF2和SRP68优先结合ac4C修饰的寡核苷酸(图5F)。随后,为了确定eEF2或SRP68是否参与HMGB2翻译的调控,首先通过瞬时siRNA介导的eEF2和SRP68敲低来评估HMGB2的表达。结果发现,eEF2敲低后HMGB2蛋白显著下调。此外,RNA下拉实验进一步证实,eEF2确实对ac4C修饰的RNA显示出更高的结合能力(图5G)。这表明eEF2是ac4C乙酰化mRNA的特异性结合蛋白。此外,内源性eEF2的RNA免疫沉淀(RIP)显示,eEF2结合的RNA中ac4C修饰显著富集。此外,NAT10的耗竭相应减少了eEF2在80S核糖体和多核糖体中的分布,这支持了eEF2作为ac4C识别器的观点。为了验证eEF2是否参与HMGB2 mRNA的ac4C乙酰化,使用RIP-qPCR研究了HMGB2 mRNA与eEF2之间的相互作用。数据显示,eEF2在HMGB2 mRNA中显著富集,而在NAT10缺陷细胞中,这种相对富集被显著抑制(图5H)。NAT10的过表达导致HMGB2蛋白表达上调,而eEF2敲低后这种上调被显著减弱。这些发现表明eEF2参与了ac4C修饰调控的HMGB2表达。
使用ac4C抗体对有无NAT10缺陷的肝癌细胞中的片段化RNA进行了免疫沉淀(图5J)。结果表明,ac4C主要富集在HMGB2的外显子1/2/3和5′UTR,而不是3′UTR。有趣的是,NAT10缺陷细胞中,外显子2、外显子3和5′UTR的ac4C富集显著降低,而外显子1则没有观察到类似效应(图5J)。HMGB2 5′UTR-报告基因荧光素酶测定结果显示,对于野生型和突变型5′UTR构建体,NAT10缺陷细胞与对照组之间在翻译上没有显著差异。研究人员构建了一个仅包含野生型HMGB2 CDS的表达构建体,以及在CXX富集区引入C到T的突变,构建了三种突变型HMGB2 CDS变体(突变体1/2/3),在所有三种突变体中,HMGB2蛋白水平几乎完全恢复(图5K)。相应地突变了pmirGLO-HMGB2荧光素酶报告基因,并进行了双荧光素酶测定,结果显示,所有三种突变体都减弱了NAT10对HMGB2翻译的抑制作用(图5L),这表明HMGB2外显子2和外显子3中的乙酰化是ac4C调控HMGB2 mRNA翻译的关键位点。为了验证eEF2是否参与HMGB2 mRNA中已确定的三个ac4C乙酰化位点,进行了RNA免疫沉淀-定量PCR(RIP-qPCR)来探究HMGB2 mRNA与eEF2之间的相互作用。RIP-PCR的结果显示,eEF2主要与HMGB2 mRNA中已确定的乙酰化CDS区域相互作用,而不是5′UTR区域(图5M)。这一相互作用通过RNA下拉实验得到了进一步证实,该实验验证了eEF2与HMGB2乙酰化CDS区域的结合(图5N)。这些数据强烈表明,eEF2可能是肝癌细胞中ac4C诱导的HMGB2 mRNA翻译的关键因子。
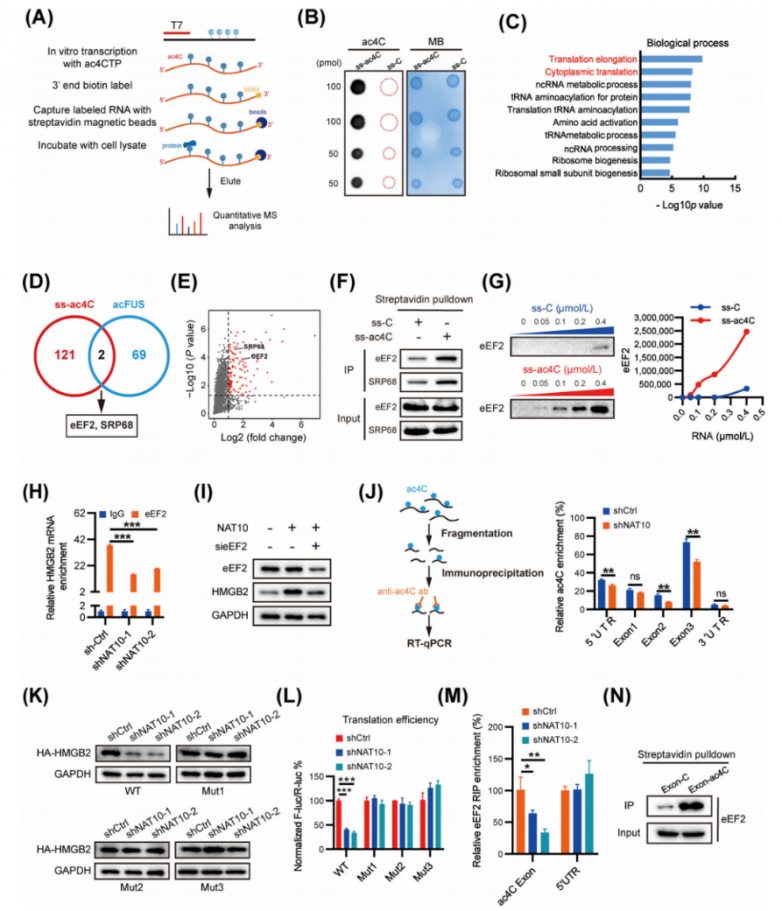
图5.HMGB2 mRNA的CDS ac4C位点增强了eEF2的结合
6.帕比司他(Panobinostat)作为NAT10介导的ac4C抑制剂的鉴定与表征
研究人员构建了表达野生型NAT10或其催化突变体(G641E)的质粒,已知该突变体会损害NAT10的RNA乙酰转移酶功能。分析结果显示,与野生型细胞相比,催化突变体NAT10的细胞中ac4C水平显著降低(图6A)。重要的是,研究人员发现NAT10的ac4C催化活性域(G641)对于其促进HCC细胞增殖和侵袭的作用至关重要。此外,催化突变体NAT10的过表达显著抑制了蛋白质合成。同时,催化突变体NAT10的过表达对HMGB2蛋白水平或mRNA ac4C水平没有影响(图6A)。研究人员基于结构的虚拟筛选、NAT10的催化口袋、抗肿瘤活性等方法选择了帕比司他作为NAT10抑制剂(图6B)。对接模型表明,帕比司他紧密结合NAT10蛋白并阻断其催化口袋(图6C)。随后,表面等离子共振分析(图6D)显示帕比司他与NAT10之间具有高亲和力(KD = 8.544 µM)。此外,帕比司他处理导致NAT10蛋白的热稳定性发生显著变化(图6E),表明其能够与NAT10结合。药物亲和力响应靶标稳定性(DARTS)试验进一步证实了它们之间的直接相互作用(图6F)。点印迹分析表明,帕比司他处理以剂量依赖的方式显著降低了总RNA和mRNA中的全局ac4C丰度(图6G-H)。此外,帕比司他处理显著抑制了蛋白质合成并抑制了HMGB2的表达(图6I)。而且,帕比司他在肝细胞癌细胞中导致HMGB2 mRNA的ac4C水平(图6J)和HMGB2翻译效率(图6K)显著降低。此外,富含在HMGB2 mRNA中的eEF2在帕比司他处理的细胞中受到显著抑制(图6L)。这些发现共同表明,帕比司他表现出对NAT10催化口袋的选择性结合和占据,从而抑制靶标RNA转录本中的ac4C修饰。
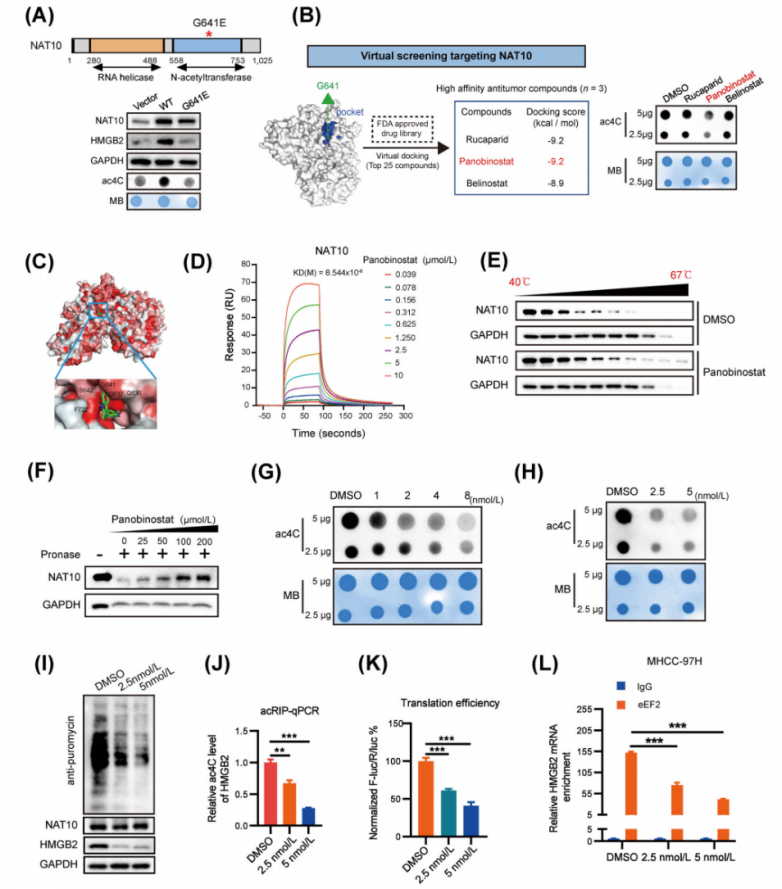
图6.NAT10抑制剂帕比司他(panobinostat)的特征
7.帕比司他在体内和体外均表现出抗肝癌活性
帕比司他显示出对MHCC-97H细胞生长的剂量依赖性抑制作用(图7A),其半数抑制浓度(IC50)为4.469 nmol/L。与NAT10敲低一致,帕比司他显著抑制了细胞增殖和侵袭(图7B-C)。随后,体内评估了帕比司他的抗肝癌作用(图7D),发现其并未引起体重减轻。在皮下异种移植模型中,帕比司他以剂量依赖的方式有效抑制了异种移植肿瘤的生长(图7E)。为了进一步验证NAT10介导的ac4C在体内的抑制作用,研究人员分别使用点杂交和Western blot检测了经帕比司他处理和对照小鼠异种移植肿瘤组织中的RNA ac4C水平和HMGB2蛋白表达。与对照小鼠相比,经帕比司他处理的小鼠RNA中的ac4C水平显著降低(图7F)。此外,帕比司他有效抑制了小鼠体内HMGB2的蛋白表达(图7G)。组织病理学检查进一步证实了帕比司他的疗效,表明其对体内NAT10和HMGB2表达的抑制作用(图7H)。此外,与对照组相比,经帕比司他处理的组肝内肿瘤结节的体积和数量均显著减少(图7I)。值得注意的是,与对照组相比,生物发光成像和肺转移病灶定量结果表明,帕比司他治疗显著抑制了肝癌细胞的肺转移(图7J)。综上所述,这些数据表明帕比司他是针对NAT10介导的ac4C的一种有效且安全的先导化合物,具有潜在的肝癌治疗价值。
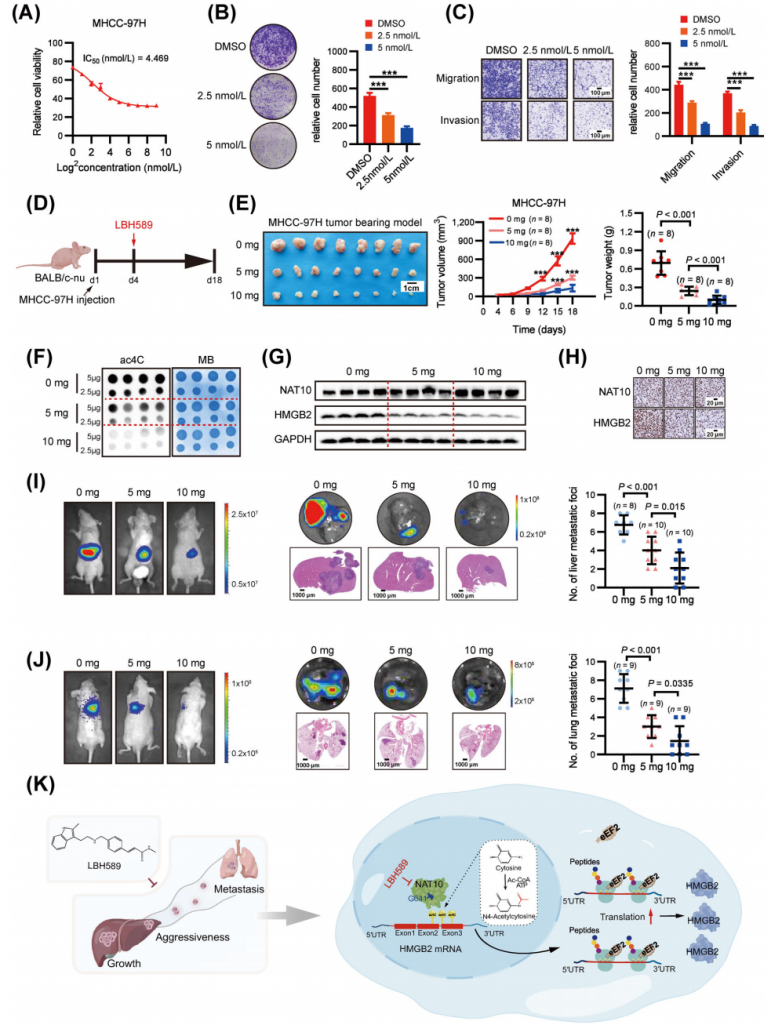
图7.帕比司他在体外和体内均显示出良好的抗HCC疗效
研究结论
该研究确定了NAT10介导的ac4C修饰触发了肝癌(HCC)中HMGB2的异常上调。作为NAT10的功能靶点,它刺激HMGB2 mRNA编码序列(CDS)内的ac4C修饰,从而增强HMGB2的翻译。此外,该研究还揭示了eEF2作为ac4C修饰mRNA的新型阅读器,展示了其与HMGB2 mRNA CDS内ac4C位点的结合并促进HMGB2的翻译。值得注意的是,该研究已经确定了帕比司他作为一种强效的NAT10-ac4C抑制剂,可有效阻止肝癌的进展。综上所述,该研究阐明了NAT10介导的mRNA ac4C修饰在肝癌发展中的关键作用,并强调了NAT10/ac4C作为肝癌治疗靶点的治疗潜力。